Alzheimer Infantile : qu’est-ce que la maladie de Niemann-Pick type C ?
 Publication validée par
Dr Marie Vanier Directeur de Recherche honoraire à l’Inserm
Publication validée par
Dr Marie Vanier Directeur de Recherche honoraire à l’Inserm
La maladie de Niemann-Pick type C provoque des troubles neurologiques graves et évolue fréquemment vers une démence. Si l'âge des cas les plus typiques lui ont valu le surnom d’"Alzheimer infantile" aux USA, la maladie fait en réalité des victimes dans toutes les tranches d’âge
Maladie de Niemann-Pick type C : une maladie neurologique avec manifestations viscérales associées
Cette pathologie génétique du métabolisme est rare : on dénombre environ 1 cas pour 100 000 naissances. En France, il y a environ une centaine de personnes touchées.
"Il s’agit d’une maladie neuro-viscérale. C’est-à-dire que nous observons chez les patients une atteinte viscérale (foie, rate, éventuellement les poumons), mais ce sont les troubles neurologiques progressifs qui en font toute la gravité", explique le Dr Marie Vanier, Directeur de Recherche honoraire à l’Inserm.
La spécialiste ajoute : "en effet, à quelques rarissimes exceptions près, le pronostic vital des patients n’est pas corrélé aux signes viscéraux, mais à l’âge d'apparition des symptômes neurologiques et à leur sévérité".
Le Dr Marie Vanier précise : "on a longtemps considéré la maladie de Niemann-Pick type C comme une affection pédiatrique, mais ce n’est pas vrai. Elle peut aussi survenir chez l’adulte. En France, on diagnostique actuellement presque autant de sujets adultes que d'enfants atteints de cette maladie".
"La plupart de mes collègues et moi-même considérons qu'appeler cette pathologie un Alzheimer de l'enfant est abusif. Ce surnom est utilisé (surtout aux USA) car c’est "vendeur". Globalement, les patients certes évoluent vers une démence, mais ce n’est pas une maladie d'Alzheimer, même s'il existe des similitudes, y compris une élévation de la protéine Tau et d'autres altérations de la pathologie cérébrale".
NPC1 et NPC2 : les deux gènes responsables
L’experte de la maladie de Niemann-Pick type C indique "cette maladie génétique peut concerner deux gènes : NPC1 ou NPC2 (soit l’un, soit l’autre)". Dans 95% des cas, la mutation est située sur le gène NPC1, et chez 5% des malades, sur NPC2.
Les mutations génétiques de ces gènes - situés respectivement sur les chromosomes 18 et 14 - provoquent un déficit des protéines qui portent les mêmes noms.
En temps normal, les protéines NPC1 et NPC2 agissent de manière concertée pour faciliter le transport du cholestérol entré dans la cellule via des lipoprotéines. Tout se passe au niveau du lysosome, structure cellulaire chargée de dégrader les molécules complexes pour en recycler les composants. Le cholestérol libéré de la lipoprotéine se lie à la protéine NPC2, qui le transfère à la protéine NPC1, cette dernière lui permettant de sortir du lysosome, afin d'être réutilisé par la cellule.
Chez les patients atteints de la maladie de Niemann-Pick type C, l’une de ces deux protéines ne fonctionne pas. "Le cholestérol s’accumule donc dans le lysosome, ainsi que d’autres lipides complexes par effet domino. Cela entraîne une cascade de réactions pathologiques au niveau cellulaire puis tissulaire, qui, à terme vont provoquer les troubles neurologiques et viscéraux que nous observons", précise l'experte.
Maladie de Niemann-Pick type C : les différentes formes
 on helping hand support and aged wood for genetic disorder awareness and children's rare disease illness")
Les symptômes de la maladie de Niemann-Pick type C appartiennent à deux catégories distinctes : viscérale et neurologique. "Les signes viscéraux apparaissent avant les signes neurologiques. Ils sont utiles, car ils peuvent aider le médecin à faire le diagnostic", précise le Dr Marie Vanier. À noter que les symptômes neurologiques peuvent apparaître beaucoup plus tardivement, et que leur âge d'apparition définit les différentes formes de la maladie.
Signes viscéraux
La maladie peut se déclarer dès la naissance, voire in-utero. L’un des signes caractéristiques chez 20 à 30% des cas est une jaunisse néonatale persistante. "Avoir une jaunisse à la naissance est fréquent. Le trouble disparaît généralement grâce à la lumière bleue au bout d’une dizaine de jours. En cas de Niemann Pick type C, elle se prolonge au-delà d’un mois, et le plus souvent, régresse vers 4/6 mois". Elle s'accompagne généralement d'une grosse rate et d'une hépatomégalie (hypertrophie du foie), qui peuvent aussi apparaître plus tard dans l'enfance, et persister, en l'absence de jaunisse néonatale antérieure, et sans manifestations cliniques particulières.
Les formes neurologiques
Chez l'enfant
"Dans le cas d’une forme infantile précoce, le développement moteur de l’enfant ne se fait pas correctement. Par exemple, il ne peut pas s'asseoir à 9 mois. Il y a aussi une hypotonie (baisse de la tonicité musculaire). Certains enfants n’apprennent jamais à marcher. Il y a aussi un retard du développement mental, mais il est moins évident que le retard moteur".
La maladie évolue rapidement chez ces formes précoces. "Ces enfants vivent rarement au-delà de 7 ans. C’est une forme très sévère, qui représente près de 20% des cas en France", prévient l’experte.
Lorsque les symptômes neurologiques apparaissent vers 3 à 5 ans, on parle de forme infantile tardive. Si c'est dans la période située entre 5-6 ans et 12-13 ans, il s'agit d'une forme juvénile. Ces formes sont beaucoup plus lentement évolutives.
Dans la forme infantile tardive, fréquente en France, ce sont le plus souvent des troubles de la marche qui attirent l’attention des parents ou des médecins. "Ces enfants ont des difficultés à apprendre à descendre les escaliers, une grande maladresse ou encore un retard de langage", détaille la spécialiste. À l’entrée à l’école, on remarque des difficultés pour apprendre à écrire et à calculer.
Les troubles d'apprentissage scolaire, des difficultés à courir ou des troubles comportementaux marquent aussi l'entrée dans la maladie des formes juvéniles.
À l'examen neurologique, on observe une ataxie cérébelleuse (perte de la coordination des mouvements musculaires lors des mouvements volontaires). Certains malades présentent une perte de tonus brusque provoquée par les émotions ou le rire (appelée cataplexie gélastique). L'apparition d'une épilepsie n'est pas rare, et l'évolution se fait vers une régression mentale progressive.
Chez l'adolescent et l'adulte
"La maladie de Niemann Pick type C peut aussi commencer à l’adolescence, d'abord avec des signes qui passent plus ou moins inaperçus : par exemple des petites difficultés d’apprentissage ou de comportement, ou d'équilibre, et ensuite une évolution très lente.
L'âge de début chez l'adulte est extrêmement variable et peut aller jusqu'au-delà de la cinquantaine. Les troubles de l'équilibre et des mouvements fins (ataxie) sont pratiquement toujours présents. Il peut aussi y avoir aussi des troubles du mouvement. Les troubles psychiatriques sont beaucoup plus fréquents et plus importants que dans les formes infantiles, et parfois au premier plan".
Les signes communs et l'évolution
La paralysie supranucléaire du regard dans le sens vertical est un signe caractéristique, observé chez la grande majorité des patients. "Il faut pour le médecin savoir rechercher ce signe, pas toujours net au début, et très utile pour suspecter le diagnostic. À terme, les parent remarquent parfois que pour regarder vers le haut ou le bas, les malades bougent la tête au lieu des yeux”, explique le Dr Vanier.
Au cours de l'évolution, l'ataxie devient de plus en plus prononcée, entraînant des chutes, et une difficulté croissante à marcher, au point de devoir utiliser un fauteuil roulant. La parole devient lente et scandée : "elle est de plus en plus difficile pour le patient qui finit par ne plus parler". Certains patients sont susceptibles de souffrir aussi de troubles de l’audition. "Ils ne sont pas sourds, mais entendent moins les sons les plus aigus".
Les problèmes de déglutition sont également très fréquents. Ils peuvent conduire à des difficultés à se nourrir (pouvant être traitées par la pose d’un bouton de gastrostomie), mais aussi des fausses routes (et par effet domino des infections pulmonaires).
L’évolution des troubles neurologiques vers la démence est fréquente, mais très variable d’un patient à l’autre. Les troubles moteurs sont souvent dominants.
La cause de décès des patients atteints de cette maladie génétique rare est très souvent neurologique ou consécutive à une surinfection pulmonaire.
"Alzheimer infantile" : diagnostic et traitements

"Concernant le diagnostic, nous avons fait de gros progrès depuis 2012-2015. Auparavant, il était nécessaire de réaliser un prélèvement de peau et on cherchait à mettre en évidence la surcharge en cholestérol dans les cellules en culture. Il fallait 2 mois pour obtenir un résultat. Puis on recherchait les mutations génétiques. Maintenant, des dosages spécifiques réalisés sur une simple prise de sang permettent, s'ils montrent une anomalie, de passer rapidement à l'étude des 2 gènes.
Malheureusement, la maladie de Niemann Pick type C est encore aujourd’hui incurable. Toutefois, une bonne prise en charge améliore considérablement la qualité de vie des patients et on peut dans une certaine mesure ralentir l'évolution de la maladie et ainsi gagner de précieuses années. On peut traiter certains symptômes, tels que l’épilepsie, cataplexie, problèmes de déglutition, surinfections pulmonaires, etc...
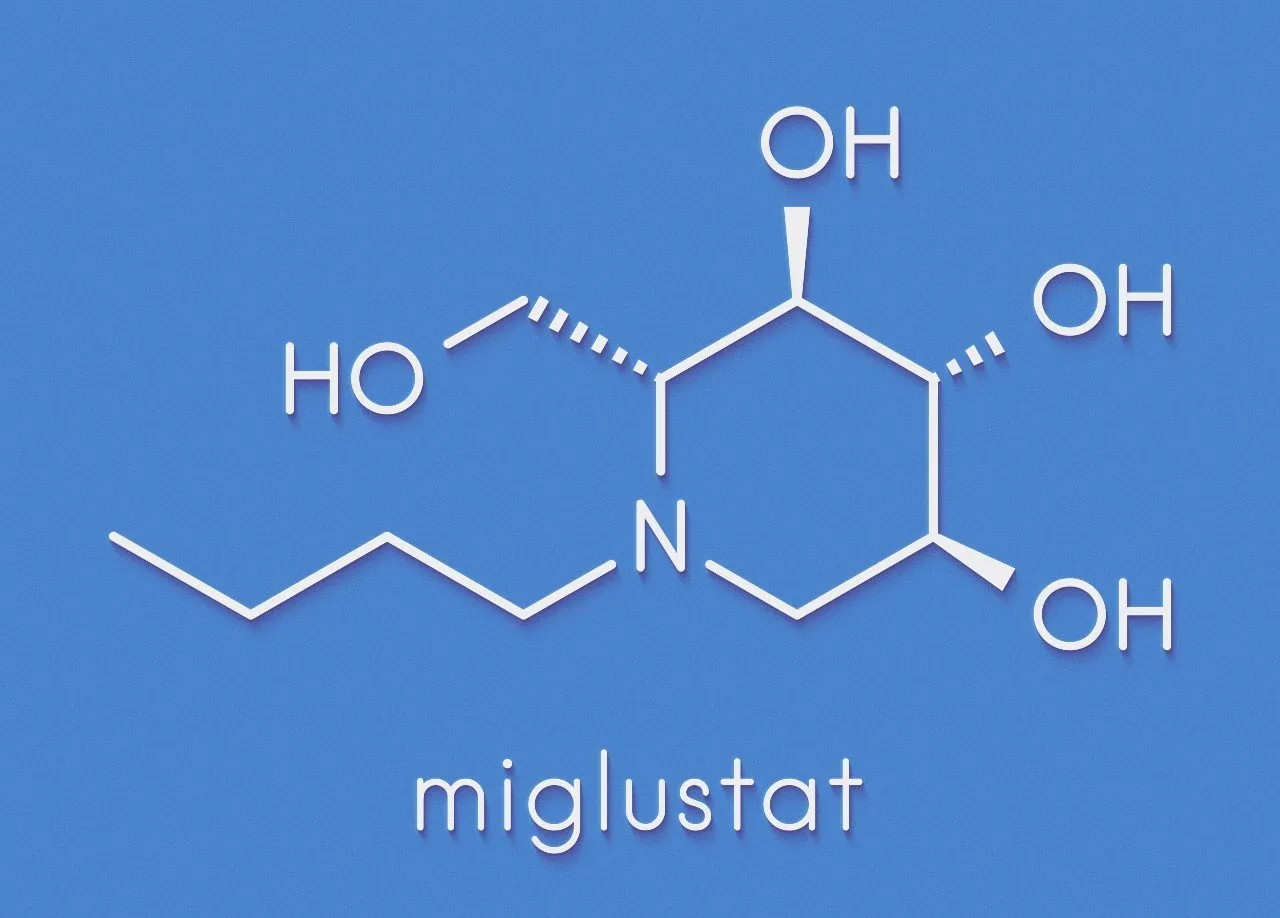
Pour l'instant, un seul médicament spécifique, le miglustat, a une autorisation de prescription. "Il n’a pas d’effet sur les signes viscéraux, mais il permet d’améliorer certains symptômes neurologiques, en particulier les problèmes de déglutition. Il ne s’agit pas d’un médicament miracle, mais certains patients peuvent être stabilisés pendant plusieurs années", précise le Dr Marie Vanier. En revanche, même si des génériques sont maintenant disponibles, ce médicament est très coûteux, ce qui en limite l'emploi dans certains pays.
Formule de la molécule du miglustat
Entre les troubles neurologiques, gastriques, moteurs, psychologiques et psychiatriques, la prise en charge doit être multidisciplinaire et associer aux divers médecins spécialistes les kinésithérapeutes, psychologues, nutritionnistes, etc…
Enfin, l’association ombrelle Vaincre les Maladies Lysosomales peut aussi apporter de l'information, du soutien et du réconfort aux patients et à leurs proches qui font face à cette maladie génétique très rare.
twitter.com/SanofiGenzymeFR/status/1231935748590514177
Trois molécules en essai clinique
Les chercheurs se sont intéressés à l'hydroxypropyl-bêta-cyclodextrine, un dérivé de la cyclodextrine. Les cyclodextrines sont très utilisées. On les retrouve par exemple dans le Febreze ou encore dans des cosmétiques, elles ont ainsi l’avantage d’être facilement accessibles. Il existe des formes de qualité pharmaceutique.
"Toutefois, l’essai clinique par voie intrathécale (par voie intrarachidienne) est en stand-by. La difficulté avec les maladies rares est que les traitements sont testés sur des très petits groupes - dans ce cas 50 patients à travers le monde - nous ne sommes donc pas à l’abri de biais liés au tirage des groupes, ou au choix des tests d'évaluation. Ici, les patients ayant reçu le médicament n'ont pas évolué durant la période de l'essai (ce qui était souhaité), mais les patients témoins non plus, contrairement à ce qui est habituel. Il faudra donc de nouvelles évaluations pour juger des effets de la molécule". Un essai parallèle avec administration par voie intra-veineuse a aussi débuté.
Une autre étude repose sur l'arimoclomol. Cette petite molécule induit dans les cellules une production accrue de composés qui, entre autres, régulent le contrôle de qualité au niveau de la chaîne de production des protéines. Elle a pour but de stabiliser les protéines mutées. "Les résultats présentés dans des congrès internationaux montrent une différence significative dans la pente d’évolution des patients traités par rapport aux non traités".
L’experte ajoute "le troisième médicament en cours de test est la N-acétyl-L-leucine. Il agirait surtout sur l’ataxie (manque de coordination fine des mouvements volontaires)".
La thérapie génique pourrait-elle être une solution ?
Le chemin sera encore long, selon la Dr Marie Vanier. "Il y a de nombreuses recherches, mais c'est compliqué du fait des propriétés de la protéine NPC1, qui ne peut pas passer d'une cellule à l'autre. Si tous les patients souffraient d’une mutation du gène NCP2, cela serait sûrement plus facile à mettre au point, car il s’agit d’une molécule soluble. Pour les patients NCP1 il faudrait toucher un maximum de neurones, en particulier dans le cervelet", prévient la spécialiste.
Toutefois, elle ne ferme pas complètement la porte. "Il y a deux ans, j’aurais dit : “oubliez”, mais je suis moins catégorique aujourd’hui, au vu des avancées technologiques. Pour la plupart des spécialistes, le traitement futur de la maladie de Niemann-Pick C combinera plusieurs approches ayant des modes d'action complémentaires, dont pourquoi pas aussi une thérapie génique ciblée, associée à diverses molécules médicamenteuses".
Afficher les sources de cet article
Merci au Dr Marie Vanier pour son aide.
L'association Vaincre les Maladies Lysosomales